

Protein Folding: An A.I. Teaching Module
Introduction
Proteins are the pillars of life. From biological catalysts to neuromodulators, protein interactions could represent, in the AI world, the black box of molecular biology. Science students often learn the main four macromolecules- lipids, carbohydrates, proteins, and nucleic acids as one of the first topics of their biology course. However, only a few students acquire a deep understanding of how these molecules shape living organisms. Molecular interactions are taught in chemistry while molecules are only presented as blurbs in most biology textbooks (see figure 1), hindering students to make clear connections between the chemical reactions learnt in chemistry and the critical role of molecular interactions in a physiological context.
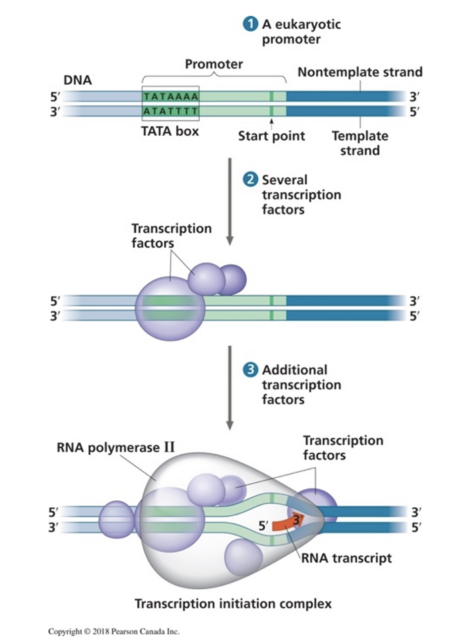
Figure 1. Representation of RNA, proteins, and enzyme (RNA polymerase), from a popular biology textbook: Campbell Biology 3rd Canadian edition.
In the last 70 years, we have deciphered how genes and DNA code for specific polypeptides and we can predict the chain of amino acid that will be produced based on a DNA sequence. However, our capacity to predict the three-dimensional structure of a polypeptide based on its basic components was, until recently, very limited. The tools available were slow and costly. Confirmation of a 3-D structure often requires X-ray crystallography, the method used by Rosalind Franklin’s team to generate photo 51 leading to the discovery of DNA 3-D structure in the 1950s.
In the past 5 years, AI-based methodologies improved the speed and precision of these predictions, impacting fundamental and medical research drastically.
This pedagogical module offers an introduction to the molecular basis of polypeptide production and folding, allowing a better understanding of the crucial impact AI-based tools will have on the future of protein folding prediction models.
Pedagogical Module
First, fundamental biochemical concepts about protein production and structure will be explored. We will then demonstrate AI contribution to the field and introduce open-source tools and software.
The module is designed as three independent activities (Part 1, 2 and 3) that can be used independently or as one single integrated activity. Parts I and II allow students to review/integrate concepts of molecular basis of protein production and folding, and part III introduces AI tools and contribution, as well as additional references/resources/games to further explore this aspect.
- The module is designed with the understanding that students have prior knowledge on protein structure and the basic concepts of cellular protein synthesis. As preparation or review, the following websites are good first-stop resource for students:
https://www.nature.com/scitable/topicpage/protein-structure-14122136/#
Part I
- The central dogma: review how DNA codes for proteins. Using the genetic code (see figure 2), students will apply coding concepts from their programming course to design an algorithm where a series of DNA codons in a gene will compute the specific amino acid sequence in a protein (primary structure). Collaboration between biology teachers and computer sciences teachers is encouraged here. Alternatively, students can do the activity on a white board or on a paper handout.
Central Dogma

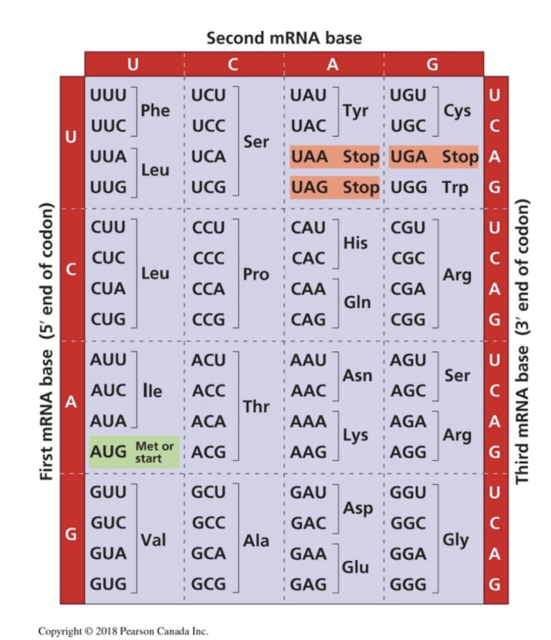
Figure 2. The genetic code states the correspondence of each RNA codon (three-letter sequence) to a specific amino acid, as deciphered by Nirenberg in 1966. From Campbell, 3rd Canadian edition.
To align with the following activities, students will use the following DNA code, which will correspond to the primary structure of the protein studied in the second and third part of the activity.
Instructions to students
- Using the following DNA coding sequence, either 1- program or 2- write the mRNA sequence formed after transcription. From the mRNA sequence, find the primary structure of the polypeptide formed after translation. For simplicity, ignore RNA modifications such as splicing.
DNA coding sequence (could be modified for the template sequence for more advanced students):
5’ ATG-CGT-TGG-CAA-GAA-ATG-GGT-TAT-ATT-TTT-TAT-CCT-AGA-AAA-TTA-CGT-TGA 3’
Transcription
mRNA:
Answer: 5’ AUG-CGU-UGG-CAA-GAA-AUG-GGU-UAU-AUU-UUU-UAU-CCU-AGA-AAA-UUA-CGU-UGA 3’
Translation
Polypeptide’s primary structure:
Answer: Met(M)-Arg(R)-Tryp(W)-Gln(Q)-Glu(E)- Met(M)-Gly(G)- Tyr(Y)-Ile(I)-Phe(F)- Tyr(Y)-Pro(P)- Arg(R)-Lys(K)-Leu(L)- Arg(R)
Part II
- Based on the polarity and charges of each amino acid (see figure 3), students will apply the concepts learnt in chemistry to predict amino acid interactions. Student should review concepts of hydrophobicity, hydrophilicity, bonds, and if possible lowest energy sate of a molecule.
The following article can be interesting to more advanced students:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC21612/
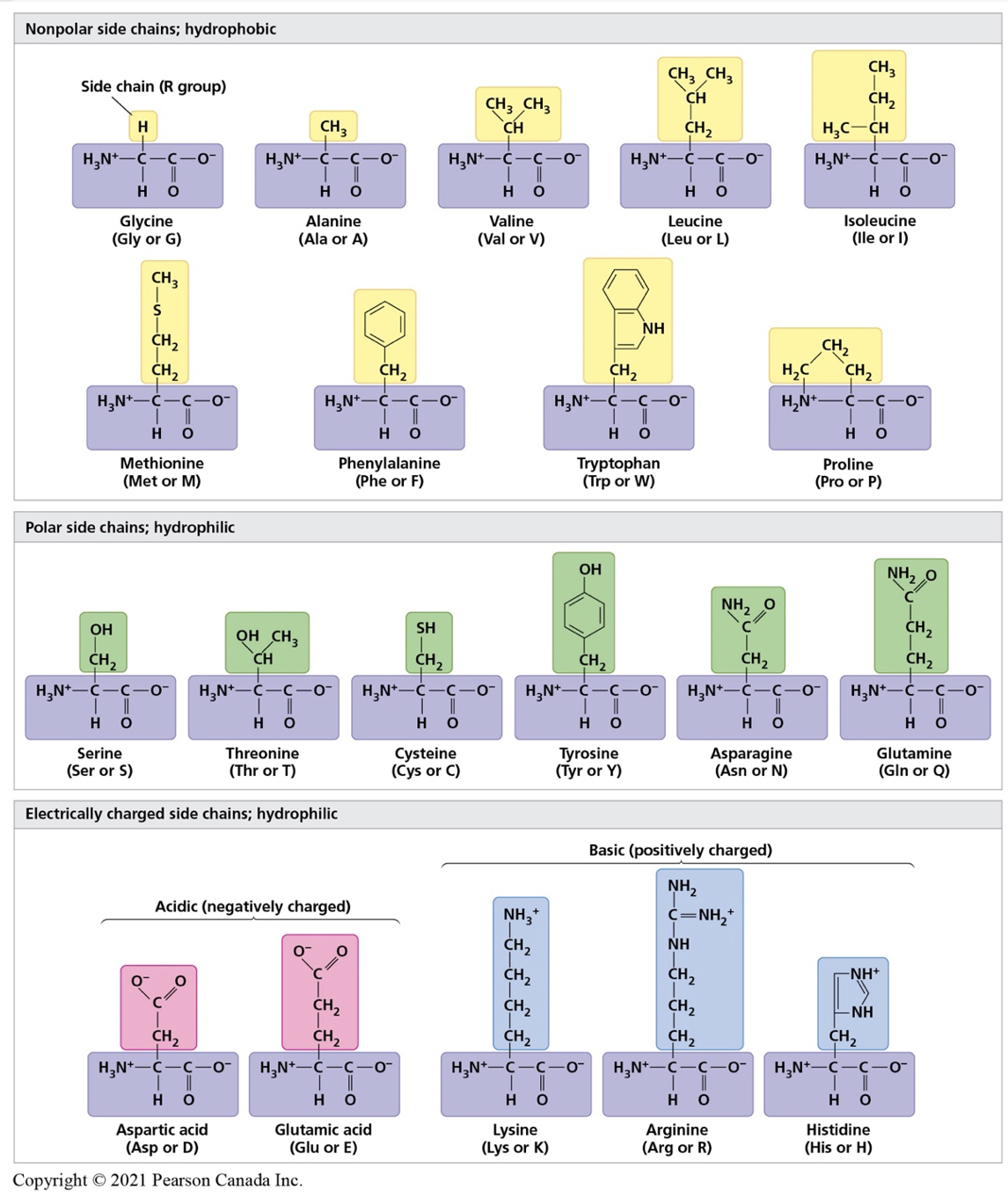
Figure 3. Properties of the 20 amino acids used to create proteins. Image from Campbell 3rd Canadian edition.
To align with the other parts of the activity, students can use the protein primary sequence presented to predict the tertiary structure of a simple protein. Teacher should print the amino acid provided, in black and white, ideally plasticised with a small hole on each side of the carboxyl and amino end, to attach a string.
Amino acids to print for students
Teachers should have post it of different colour available (green, yellow, red and blue) as well as string and scissors.
Instructions to students:
- Before beginning the activity, discuss with your team why it is important to the scientific community to understand and predict protein structure. The following article can inform your discussion: https://www.nature.com/articles/s41586-021-03819-2
- Amino acids forming the primary structure of a polypeptide are provided (see below). Examine the R group of each of the amino acid and predict the chemical property of each. Write your result in pencil in table 1. Discuss with your team and teacher and confirm your answers with figure 3.
-
- Mark each amino acid provided with its correct property. Use the colour code below and the post-it provided to identify each amino as (at physiological pH of 7.4):
- Polar (green), nonpolar (yellow), positively charged (red), or negatively charged (blue)
3. Using the material provided, attach each amino acid listed to the next one through covalent bonds (knots!). The amino acids should be attached on a string in the order presented, to represent the primary structure of a polypeptide.
Polypeptide primary structure:
Met(M)-Arg(R)-Tryp(W)-Gln(Q)-Glu(E)- Met(M)-Gly(G)- Tyr(Y)-Ile(I)-Phe(F)- Tyr(Y)-Pro(P)- Arg(R)-Lys(K)-Leu(L)- Arg(R)
4. Fold your polypeptide into a possible secondary, and then tertiary structure molecule. Fold the string (protein) in a tertiary structure that would respect the basic chemical and physical constrains: positively and negatively charged amino acid will attract each other, non-polar amino acid will be located toward the inside of the protein, etc. More than one answer is possible.
For an overview on protein folding and protein folding dynamics, consult the following articles:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7545034/figure/F2/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7545034/
- https://bio.libretexts.org/Bookshelves/Biochemistry/Fundamentals_of_Biochemistry_(Jakubowski_and_Flatt)/01%3A_Unit_I-_Structure_and_Catalysis/04%3A_The_Three-Dimensional_Structure_of_Proteins/4.08%3A_Protein_Folding_and_Unfolding_(Denaturation)_-_Dynamics
-
- For each amino acid in the polypeptide’s primary structure, note the polarity/charge of each R group
Table 1. Polarity of various amino acids based on the R group.
| 1. Methionine (M): | 2. Arginine (R): |
| 3. Tryptophan(W): | 4. Glutamine(Q) |
| 5. Glutamic Acid(E): | 6. Glycine(G): |
| 7. Tyrosine(Y): | 8. Proline(P): |
| 9. Lysine(K): | 10. Leucine(L): |
ANSWER KEY:
| 1. Methionine: Non polar | 2. Arginine: Positively charged (basic) |
| 3. Tryptophan: Non polar | 4. Glutamine: Polar |
| 5. Glutamic Acid: Negatively charged (acidic) | 6. Glycine: Non polar |
| 7. Tyrosine: Polar | 8. Proline: Non polar |
| 9. Lysine: Positvely charged (basic) | 10. Leucine: Non polar |
Part III
The last topic introduces AI software in biology and how they revolutionized the field of protein structure prediction.
First students can play with an introductory game to introduce the topic of protein evolution using multiple sequence alignment, based on sequence and structural homology.
Some teachers and students may be interested in the theory behind these models, commonly used in protein structure prediction: https://www.sciencedirect.com/science/article/pii/B9780323897754000237
Homology modelling:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/homology-modeling
Phylo:
“The comparison of the genomes from various species is one of the most fundamental and powerful technique in molecular Biology. It helps us to decipher our DNA and identify new genes. Through it may appear to be just a game, Phylo is actually a framework for harnessing the computing power of mankind to solve the Multiple Sequence Alignment problem.”
This game is developed by a team at McGill, and the programmer can open an adapted ‘course module’ for teachers interested.
Instructions to students
- In this activity, you will start with the primary structure of a simple existing protein (see part I and II) and use an open-source protein database to explore what scientists know about this polypeptide.
-
- UniProt database provides the primary structure and the predicted tertiary structure of many polypeptides. Access the site https://www.uniprot.org/
- Select Proteins-UniProt Knowledgebase
- In the search box at the top, search: MOTSC_HUMAN, and then click on the blue link that appears under Entry. The entry number is AOAOC5B5G6, the gene name is MT-RNR1 (see link for advanced students https://www.aging-us.com/article/102944/pdf or https://pubmed.ncbi.nlm.nih.gov/25738459/ )
- Using the titles on the left, get familiar with your protein:
- What is the Recommended name of the Protein? ____________________________________
(answer: Mitochondrial-derived peptide MOTS-c)
-
- Where is it found (subcellular location or destination)
___________________________
___________________________
___________________________
(answers: secreted, mitochondrion, nucleus)
2. Now click on Structure and look at the predicted structure.
Click on the image generated to zoom and change the view angle. Double-click on an amino acid to view its molecular structure. Compare with your predicted structure in part II.
*Note that this is an extremely short protein, hence the tertiary structure is simpler and not representative of a complex protein with sophisticated secondary and tertiary structure. As a simpler exercise, go back to the search and look for human insulin:
INS_HUMAN.
Congratulations, you are now acquainted with the world of protein folding and some associated technological tools.
It is now time for you have fun and explore, it requires time and patience to get a good understanding of the many forces at play when it comes to protein folding, and even Chat-GPT is not able to help!
- You are now ready to explore some protein folding games (be careful, it is addictive!).
Go to the Fold it web site and download the app. https://fold.it/. You can now explore the educational side or go directly to the gamer’s world.
You can also explore the protein folding predictor that changed the scene: Deep Mind’s Alpha Fold https://alphafold.ebi.ac.uk/
Interesting fact: An independent organization plans a bi-yearly ‘contest’ to provide a mechanism for the assessment of methods of protein structure modelling. It is interesting to see the leap taken when Alpha Fold’s new tool was introduced. Check out their website ‘Critical Assessment of Techniques for Protein Structure Prediction- CASP’: https://predictioncenter.org/casp15/index.cgi
References:
Campbell Biology, 3rd Canadian Edition, 2021. Cain, M.L., Durnford, D.G., Minorsky, P.V., Moyes, C.D., Rawle, F.E., Reece, J.B., Scott, K., Urry, L.A., Wasserman, S

